
第五章 追獵化學失衡 (p. 101)
科學的大悲劇,便是以醜陋的事實扼殺美麗的假說。
——湯瑪士·赫胥黎(Thomas Huxley, 1870) (^1)
成人的大腦重約1.5公斤,把它從頭骨挪出來,靠近點看它會比你想像的還大一些。我一度以為人腦能夠輕易地放在手掌心,但事實上你需要用兩手才能將它穩穩捧起來。如果這顆大腦是新鮮的,還沒被泡進福馬林做成標本,表面會有蜘蛛網般、粉色的血管網絡,而且腦組織摸起來軟軟的,像凝膠。人腦的性質的確是「生物學的」,然而它也以某種方式造就了人類心靈所有不可思議且非凡的才能。因為我的一位朋友,麻州總醫院神經科學家車章浩(Jang-Ho Cha,音譯)的邀請,我參加了該院的腦部切片研討會,當時我認為親眼見到人腦,可讓我更能想像那些據稱導致憂鬱和精神病的神經傳導路徑;但很自然的,參訪中所獲得的遠比這多得多。能近距離觀察人的腦部,著實令人屏息。
目前,我們對人腦傳訊系統的原理已有相當程度的了解。車章浩提到,人腦中有1,000億個神經元。一個「典型」神經元的細胞,會從範圍廣大的樹突(dendrites)網絡接收訊號,並經由單一的軸突(axon)傳出訊息,軸突可投射到腦內遠端的區域(或向下傳至脊髓)。軸突結束之處分支為許多末端,多巴胺、血清素等化學傳訊
者便是從這些軸突末端將訊號釋放至約20奈米寬(1奈米為10億分之1公尺)的突觸間隙(synaptic cleft)。一個神經元有1,000到1萬個突觸連結,一個成人的腦部總計約有150兆個突觸。
使用同一種神經傳導物質神經元的軸突通常會形成一束,有點像電纜線,而科學家發現多巴胺、正腎上腺素和血清素在福馬林蒸氣下會發出不同的螢光色,讓他們得以追蹤這些腦中神經傳導物質的路徑。約瑟夫·希爾德克勞特提出情感障礙症的理論時,認為正腎上腺素最有可能是憂鬱時所缺乏的神經傳導物質,但後繼的研究者們很快就把注意力轉移到血清素上。為了探究精神疾病的化學失衡理論,我們必須看一看腦中憂鬱症的路徑,以及思覺失調症的多巴胺路徑。
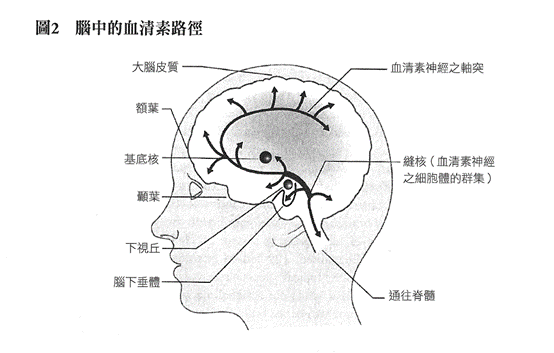
血清素路徑有著古老的演化根源。所有脊椎動物與多數無脊椎動物的神經系統內,都可發現血清素神經元,它在人類身上的細胞體位於腦幹內一處被稱為縫核(raphe nuclei)的區域。這些神經元有些向下長出長條的軸突直至脊髓,脊髓是涉及控制呼吸、心臟以及腸胃活動的系統;其他的血清素神經元則往上延伸至所有腦部區域的軸突,包括小腦、下視丘、基底核、顳葉、邊緣系統(limbic region)、大腦皮質,以及額葉。這條路徑與記憶、學習、睡眠、食慾,以及情緒和行為的調控有關。如紐約大學生物學教授艾弗蘭·埃思米迪亞(Efrain Azmitia)所言,「腦部的血清素系統是人們所知最大的單一腦部系統,可被描述為『巨大』神經系統」。(^2)
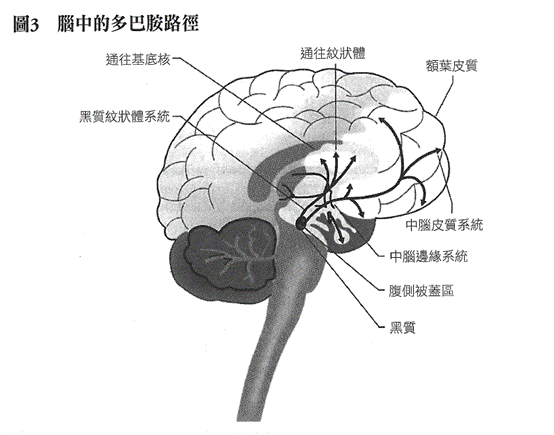
腦中有三條主要的多巴胺路徑。這三個系統的細胞體皆位
於腦幹上方,在黑質(substantia nigra)或腹側被蓋區(ventral tegmentum)。它們的軸突投射至基底核(黑質紋狀體系統)、邊緣系統(中腦邊緣系統),以及額葉(中腦皮質系統)。基底核啟動並控制運動。邊緣系統的結構,包括嗅結節(olfactory tubercle)伏隔核(nucleus accumbens)和杏仁核(amygdala),位於額葉後方,幫助調控情緒。我們就是在此處得以感受到這個世界,這是我們產生自我感與現實感至關重要的過程。額葉是人類腦部最與眾不同的區域,提供我們如上帝般的能力,得以觀照自身。
這一切生理學的知識,包括1,000億個神經元、150兆個突觸,以及各種神經傳導物質的路徑,都說明了大腦幾乎是無與倫比的複雜。可是精神疾病的化學失衡理論卻將這種複雜性簡化為簡單的、易於理解的疾病機轉。憂鬱症的問題在於血清素神經元釋出至突觸
間隙的血清素太少,因此腦中的血清素路徑「活性不足」;而抗憂鬱劑使突觸間隙的血清素濃度回到正常,使這些路徑能以適當的步調傳遞訊息。同時,思覺失調症典型的幻覺和幻聽,是源於過度活化的多巴胺路徑。若非突觸前神經元釋出過多的多巴胺至突觸,就是目標神經元具有異常高密度的多巴胺受體;抗精神病劑為這個系統踩了煞車,使多巴胺路徑能以較為正常的方式運作。
這就是希爾德克勞特和雅克.凡羅森提出的化學失衡理論也正是那份讓希爾德克勞特提出其假說的研究,為研究者提供了測試假說的方法。異菸鹼異丙醯肼和丙咪嗪的研究顯示,’/ 。血清素被代謝為5-氫氧靛基醋酸(5-Hydroxyindoleacetic acid,5-HIAA);多巴胺轉變為高香草酸(homovanillic acid,HVA)。研究者可仔細尋找腦脊髓液中的這些代謝物含量,其數值能間接測量突觸之神經傳導物質的濃度。既然理論上憂鬱症是由血清素濃度太低所導致,任何處於這種情緒狀態者腦脊髓液的5-氫氧靛基醋酸都應該低於正常值;同樣地,既然理論上過度活化的多巴胺系統會導致思覺失調症,那麼有幻聽或偏執妄想者的腦脊髓液中都應該含有異常高濃度的高香草酸。
這條研究路線讓科學家忙了將近十五年的時間。
接受考驗的血清素假說 (p. 105)
1969年,耶魯大學的麥爾坎‧鮑爾斯(Malcolm Bowers)是首位針對憂鬱症患者的腦脊髓液中是否有較低濃度之血清素代謝物提出報告的人。在一份針對8位憂鬱症患者的研究中(這些患者先前皆使用過抗憂鬱劑),他宣布患者的5-氫氧靛基醋酸濃度比正常值低,但並非「顯著」的低。(^3)兩年後,麥基爾大學的研究者也說,他們無法在憂鬱症患者與正常的控制組之間找到5-氫氧靛基醋酸濃度有「統計上顯著」的差異,亦無法找到憂鬱症狀嚴重程度與5-氫氧靛基醋酸濃度有任何關連。(^4)1974年,鮑爾斯再度進行一項調控更加精密的追蹤研究:從未使用抗憂鬱劑的憂鬱症患者具有完全正常的5-氫氧靛基醋酸濃度。(^5)
憂鬱症的血清素理論似乎無法成立。1974年,兩位賓州大學的研究者約瑟夫·孟德斯(Joseph Mendels)與艾倫·弗雷澤(Alan Frazer)重新檢視那些一開始引導希爾德克勞特推展其理論的證據。希爾德克勞特指出,消耗腦中單胺類(正腎上腺素、血清素,以及多巴胺)的蛇根鹼,通常使人們憂鬱;但當孟德斯與弗雷澤仔細檢視科學文獻發現,事實上對高血壓患者投以蛇根鹼,只有6%的人變得憂鬱。更有甚者,1955年,一群英國醫師對他們的憂鬱症患者投以這種草藥,它反而提振了許多人的精神。孟德斯與弗雷澤的結論是,蛇根鹼根本不會引發憂鬱;(^6)他們也提到,即使投以其他消耗單胺類的藥物,這些藥劑亦不會引發憂鬱。他們寫道,「此處回顧的文獻,強烈顯示出腦中單胺類的消耗,無論是多巴胺或血清素本身,皆無法充分解釋憂鬱之臨床症狀的產生」。(^7)
看來,這個理論即將被宣告死亡,並埋葬起來。但1975年,斯德哥爾摩的卡羅琳斯卡學院(Karolinska Institute)的瑪麗·艾斯柏格(Marie Asberg)與她的同事,為這個理論注入了新生命。在他們測試的68位憂鬱症患者中,有20位的5-氫氧靛基醋酸濃度較低,而這些血清素不足的患者比其他患者更具有自殺傾向,事實上這20位患者中最終有2位自殺了。這群瑞典的研究者說,這就是證據,顯示可能存在「有一個次群體的憂鬱症是生化因素所致,其特色即是血清素代謝轉換的擾動」。(^8)
沒多久,美國著名的精神科醫師便寫道有「將近30%」的憂鬱症患者血清素濃度較低。憂鬱症的血清素理論似乎至少獲得部分的平反。但今日若重新檢視艾斯柏格的研究並檢驗其資料,我們可以看到她所謂憂鬱症患者中「有一個次群體是生物因素所致」的發現,幾乎就是個一廂情願的說法。
在其研究中,艾斯柏格報告指出「正常」群組中有25%,每毫升腦脊髓液中5-氫氧靛基醋酸的含量低於15奈克(nanogram),50%的人每毫升具有15到25奈克的5-氫氧靛基醋酸含量,剩下的25%則是超過25奈克。她的「正常人」的鐘形曲線顯示,5-氫氧靛基醋酸含量的變化相當大。但她未能在討論中記錄的是,該研究中68位憂鬱症患者的鐘形曲線和正常人幾乎完全相同。這些患者中有29%(68位中有20位)的5-氫氧靛基醋酸含量低於15奈克,47%的患者含量為15到25奈克,而24%的患者有超過25奈克的含量。這29%的憂鬱症患者,在他們的腦脊髓液中可能有「低的」血清素代謝物含量(這就是她所謂的「生物因素造成的次群體」),但「正常」人也有25%是如此。正常人的中間值是20奈克,而結果卻有超過一半的憂鬱症患者(68位中有37位)的含量超過此數值。
這樣看來,艾斯柏格的研究並未提供任何新的理由使人相信憂鬱症的血清素理論。過沒多久,日本的研究者也在無意間揭露此理論背後的錯誤邏輯。他們報告指出,有些日本使用的抗憂鬱劑阻斷了血清素的受體,抑制那些路徑的啟動,因此推論憂鬱可能是由於「突觸間隙過多游離的血清素」所造成。他們所使用的反向推論,就是和推導出憂鬱症的低血清素理論一樣的方式,所以如果他們想這麼做,這些日本科學家也可以搬出艾斯柏格的研究來支持自己的理論,因為這些瑞典人也發現24%的憂鬱症患者有「高」血清素濃度。
1984年,美國國家精神衛生研究院的研究者再次研究憂鬱症的低血清素理論。他們想看看憂鬱症患者中那些具有「低」血清素濃度的「生物因素次群體」,對一種作用為選擇性阻斷血清素回收,名為阿米替林(amitriptyline)的抗憂鬱劑,是否有最佳的反應。如果抗憂鬱劑是腦中化學失衡的解藥,那麼阿米替林應該會對這個次群體有最佳的療效。但研究主持人詹姆士‧馬斯(James Maas)寫道,「與我們期待的相反,腦脊髓液的5-氫氧靛基醋酸濃度和患者對阿米替林的反應沒有關連」。此外,他與其他美國國家精神衛生研究院的研究者發現,憂鬱症患者的5-氫氧靛基醋酸濃度變化甚大,而這與艾斯柏格的發現是一樣的。有些人的腦脊髓液裡血清素代謝物的濃度高,但有些人則濃度低。美國國家精神衛生研究院的科學家只能得出一種可能的結論:「血清素系統本身活動的增加或減少,不太可能和憂鬱症有關。」(^i)
I 即便發布了這份報告,憂鬱症的血清素理論此一說法並未完全消失。禮來大藥廠於1988年帶進市面的百憂解,就是一種「選擇性血清素回收抑制劑」(selective serotonin reuptake inhibitor, SSRI),它在商業上的成功促使認為「憂鬱症是由於血清素濃度不足所致」這種國國家精神衛生研究院的研究者也探查不同的神經傳導物質濃度,與患者對抗憂鬱劑的反應之間,其他可能的關連。研究者測量正腎上腺素代謝物與多巴胺代謝物的濃度;他們將憂鬱患者分為雙相情緒障礙症與單相性憂鬱症兩組;評估患者對丙咪嗪和阿米替林這兩種抗憂鬱劑的反應。研究者在這些子群體中不少的組別,與其對兩種藥物之一的反應發現些微的關連;我在此著重的研究發現有以下幾點:(1)憂鬱是否由低濃度的血清素所導致,(2)低血清素濃度的患者子群體,是否對選擇性阻斷神經傳導物質之回收的藥物有更佳的反應。
說法再度流行起來,並再度有許多研究者開始進行試驗,測試是否真是如此。但第二回合的試驗結果跟前一回合並無不同。「我職業生涯的頭幾年整天都花在腦部血清素代謝的研究,但從未看到任何有說服力的證據能證明任何精神疾病,包括憂鬱症在內,是源自腦部缺乏血清素。」史丹佛大學的精神科醫師大衛.柏恩斯(David Burns)2003年時這麼說。(^11)其他許多研究者也提出同樣的說法。達拉斯西南醫學中心的精神科副教授柯林·羅斯(Colin Ross),在其1995年的著作《生物精神醫學中的偽科學》(Pseudoscience in Biological Psychiatry)中寫道,「沒有任何科學證據指出,臨床所見的憂鬱症是起因於任何一種生理上的匱乏狀態」。(^12)2000年,《基礎精神藥理學》(Essential Psychopharmacology)一書的作者對醫學生說,「沒有明確且令人信服的證據能證實,缺乏單胺類是憂鬱症的成因;也就是說,並沒有『真正的』缺乏單胺類這回事」。(^13)然而,在藥品廣告推波助瀾下,這種信念持續存在,而這讓著有多本精神醫學史著作的愛爾蘭精神科醫師大衛·希利(David Healy)於2005年出言嘲諷,認為此理論應該要被丟到醫學的垃圾桶裡,跟其他假的理論擺在一起。「憂鬱的血清素理論,」他的語氣中帶有明顯地惱怒,「可以跟瘋癲的手淫理論相提並論。」(^14)
多巴胺的舊事重演 (^109)
凡羅森提出思覺失調症的多巴胺假說時,提到研究者第一件需要做的事是「進一步證實」抗精神病劑確實阻礙了腦中的多巴胺傳遞。這要花一些時間,而直到1975年,約翰霍普金斯醫學院的索羅門·史奈德(Solomon Snyder)與多倫多大學的菲力普‧西曼(Philip Seeman)才為藥物如何造成效果的理論增添了內涵。首先,史奈德辨別出兩種多巴胺受體,稱作第一型多巴胺受體與第二型多巴胺受體(D, and D, receptors);接著,兩位研究員都發現抗精神病藥阻斷了70-90%的第二型多巴胺受體。(^15)此時報紙便開始報導,這些藥物可能如何矯正了腦中的化學失衡。
「腦中有太多發揮作用的多巴胺,而這能說明為何思覺失調症患者會有那些幾欲將人淹沒的豐沛情感,」《紐約時報》這麼解釋,「透過阻斷腦中的多巴胺受體,神經抑制劑能將那些並非真實存在的景象和聲音關起來。」(^16)
然而,就算史奈德與西曼提出如上的研究結果,麥爾坎‧鮑爾斯的發現仍為多巴胺假說蒙上陰影。他測量未用藥的思覺失調症患者腦脊髓液中多巴胺代謝物的濃度,發現濃度相當正常。「我們的發現,」他寫道,「無法為支持患者的中腦多巴胺系統過度活化的情形,增加神經化學方面的證據。」(^17)其他研究者很快也提出類似的報告。1975年,美國國家精神衛生研究院的羅伯特·普斯特(Robert Post),測量20位未用藥之思覺失調症患者腦脊髓液中的香草酸濃度,發現「與控制組並無顯著差異」。(^18)病理解剖的研究亦顯示,未用藥的思覺失調症患者,其腦組織的多巴胺濃度並無異常。1982年,加州大學洛杉磯分校的約翰·赫拉池(John Haracz)回顧這些研究,得出相當明顯且重點的結論:「這些研究結果並不支持(未用藥的)思覺失調症患者腦部有較多的多巴胺代謝。」(^19)
發現未用藥的思覺失調症患者有正常的多巴胺濃度後,研究者把注意力轉向第二種可能性。或許思覺失調症患者有過多的多巴胺受體。如果是這樣,突觸後神經元會變得對多巴胺「過敏」,而這會造成多巴胺路徑的過度活化。1978年,多倫多大學的菲力普‧西曼在《自然》期刊宣布確實如此。病理解剖顯示,20位思覺失調症患者腦部的第二型多巴胺受體比常人多70%。乍看之下,人們似乎找到了思覺失調症的原因,但西曼也提出警告,這些患者在死亡前皆有使用過神經抑制劑。「雖然這些研究結果大體上與思覺失調症的多巴胺假說明顯相符」,他寫道,第二型多巴胺受體的增加可能「源於長期使用神經抑制劑。」(^20)
各式各樣的研究很快證實,藥物才是兇手。對大鼠投以神經抑制劑,牠們第二型多巴胺受體的數量便快速增加;(^21)若對大鼠投以阻斷第一型多巴胺受體的藥物,這種類型的受體分布的密度就會增加。(^22)在不同的例子中,受體增加是腦部試圖彌補藥物阻斷其訊號的證據。緊接著在1982年,安格斯•麥凱(Angus MacKay)與他的英國同僚報告指出,當他們檢驗48位思覺失調症死亡患者的腦部組織,「(第二型)多巴胺受體增加的狀況,只出現於持續使用神經抑制劑至死亡的患者身上,顯示這完全是醫源性的(藥物造成的)結果」。(^23)幾年後,德國研究者指出,他們的病理解剖研究也得到相同的結果。(^24)最後,法國、瑞典,與芬蘭的研究者使用正子攝影,在活著且從未使用過神經抑制劑的患者身上,研究第二型多巴胺受體的分布密度,所有結果都顯示,思覺失調症患者與「正常控制組」之間「無顯著差異」(^25)
自那時起,研究者持續研究那些被診斷有思覺失調症者的多巴胺路徑究竟哪裡出了問題,偶爾會有人報告,在某個子群體的患者中找到了某些類型的異常之處。但到1980年代晚期,思覺失調症的化學失衡假說,即認定這是一種由過度活化的多巴胺系統所導致的疾病,且可利用藥物把它調整得平衡一些的說法,已經到達終點。1990年,皮耶·丹尼克觀察到,「思覺失調症的多巴胺理論,對精神科醫師而言僅剩些許的可信度」。(^26)四年後,長島猶太醫學中心一位知名的精神科醫師約翰·凱恩(John Kane)也附和這種想法,提到「沒有夠好的證據能證明,思覺失調症代表多巴胺功能受到任何擾動」。但社會大眾持續得到的訊息仍是,被診斷為思覺失調症者具有過度活化的多巴胺系統,而藥物就如同「糖尿病的胰島素」。因此美國國家精神衛生研究院前院長史蒂芬·海曼(Steven Hyman),在他2002年的著作《分子神經藥理學》(Molecular Neuropharmacology)中,再次提醒讀者要注意事實真相。他寫道,「沒有任何可靠的證據能證實多巴胺系統的損傷是思覺失調症的主因」。(^28)
理論的輓歌 (^112)
憂鬱症的低血清素假說,以及思覺失調症的高多巴胺假說,一直以來都是精神疾病化學失衡理論的兩大支柱,而到1980年代晚期,兩者都被發現其缺失之處。其他精神疾病同樣也是以該疾病是由化學失衡所造成的樣貌呈現於大眾面前,但從未有任何證據能支持這些主張。家長們被告知,診斷為注意力不足過動症的孩童是因為多巴胺濃度不足,但其實這麼做唯一理由是利他能會刺激神經元釋放更多的多巴胺。這成了藥廠一再仰賴的故事公式:研究者會辨別出某一類藥物的作用機制,看這些藥物是如何降低或升高腦中神經傳導物質的濃度,不久後社會大眾便會被告知,以該藥治療的患者之所以致病是因為與其相反的問題。
從科學的觀點回顧,化學失衡的假說很明顯地並不穩固,許多目睹這假說樓起樓塌的科學家回想過去時都帶著一抹尷尬。早在1975年,約瑟夫·孟德斯和艾倫·弗雷澤就下了結論,希爾德克勞特的憂鬱症假說是出自「狹隘的思維」,而之所以會有此思維,是因為「當某些研究發現與起始假設不符時,沒有對其進行適切的評估」。(^29)1990年,丹尼克認為思覺失調症的多巴胺假說也有同樣的問題。當精神醫學研究者將藥物改頭換面為「抗精神病」藥物,丹尼克提到,他們實在說得「有點超過……人們可以說神經抑制劑消除了特定思覺失調症的現象,但(此藥)不能假裝是這些精神病的病因治療」。(^30)大衛·希利在他的《精神藥理學之創建》(The Creation of Psychopharmacology)一書中寫道,精神疾病的化學失衡理論被精神科醫師擁護,是因為它替他們「創造了一個可以成為真正醫師的舞台」。(^31)內科醫師有抗生素,現在精神科醫師也能有「對抗疾病」的藥丸了。
雖然社會對化學失衡的信仰依然存在(理由後續會探討),使得那些調查並書寫這段歷史的人一再強調相同的重點結論。密西根大學的神經科學教授艾略特·華倫斯坦在他1998年的著作《歸咎於腦》一書中提出結論,「現有的證據並不支持任何精神疾病的生化理論」。(^32)美國公衛署沙契爾在他1999年的報告書《精神衛生》中也坦承,「精神疾病的確切原因(病因)仍不清楚」。(^33)哈佛醫學院的精神科講師約瑟夫.葛蘭姆倫(Joseph Glenmullen)在《百憂解的後座力》(Prozac Backlash)一書中提及,「每次人們認為找到了一種化學失衡,後來就被證明是錯的。」(^34)最後,2005年,《心理醫學》(Psychological Medicine)的共同總編輯肯尼斯·坎德勒(Kenneth Kendler)為這整個故事執筆寫下簡潔而令人欽佩的墓誌銘:「我們為精神疾病追索重大且簡單的神經化學解釋,但毫無發現。」(^35)
這帶領我們進入下一個大哉問:若精神科用藥並未修復腦中的化學異常,那它們做了什麼?
百憂解,常在我心 (p. 114)
1970到1980年代,研究者將不同種類的精神科用藥如何作用於腦部,以及腦部如何回應這些藥物的詳細記錄彙集起來。我們可以講述抗憂鬱劑、神經抑制劑、苯二氮平類藥物,或是興奮劑的歷史,而所有這些歷史會顯露出其背後共同的運作過程。在大眾的心目中,化學失衡的故事是從禮來大藥廠將百憂解(氟西汀,fluoxetine)帶進市面後才真正深植人心,那麼回顧禮來的科學家與其他研究者發表在科學期刊上的報告,看看他們是怎麼說明「選擇性血清素回收抑制劑」的確起了作用,似乎頗為適當。
如同先前提到的,一旦突觸前神經元釋放血清素到突觸間隙,就必須被快速移除,才能明確地終止信號。小部分的血清素會由一種酵素代謝掉,剩下的部分則是被送回突觸前神經元,經由一種名為血清素轉運體(serotonin reuptake transport, SERT)的通道傳送。氟西汀會阻斷這個回收的通道,禮來的科學家詹姆士‧克萊門斯(James Clemens)在1975年寫道,這會造成「血清素堆積在突觸」。(^36)
然而,一如禮來的研究者所發現的,回饋機制隨之出現。突觸前神經元末端的細胞膜具有「自體受體」(autoreceptors),能監測突觸的血清素濃度。一位科學家調侃道,如果血清素濃度太低,這些自體受體會大喊「打開血清素機器」;濃度太高則會大喊「把它關掉」。這是演化設計的回饋循環,以保持血清素系統的平衡,而氟西汀觸發的是後面那則訊息。當突觸的血清素不再被清除,自體受體會命令突觸前神經元以超級緩慢的速率啟動,神經元便會開始釋放比平常少量的血清素至突觸。
回饋機制也改變了突觸後神經元。禮來的科學家於1981年提出報告,四週內,突觸後神經元血清素受體的密度會比正常值少25%;(^37)其他研究者也隨後指出,「長期的氟西汀治療」可能導致腦部特定區域的血清素受體減少50%。(^38)因此,突觸後神經元變得對化學傳訊者「不敏感」了。
由此看來,腦部似乎已成功地適應藥物。氟西汀阻斷突觸的血清素正常回收,但接著突觸前神經元就會開始釋放較少的血清素突觸後神經元則變得對血清素較不敏感,也因而不太容易啟動。此藥是設計來促進血清素路徑,而腦部的回應則是踩下煞車。這使得血清素路徑或多或少處於平衡狀態,它是研究者稱為「突觸韌性」(synaptic resilience)的適應性反應。(^39)然而,用藥頭兩週還發生了另一項改變,最終造成腦部補償反應的短路。突觸前神經元之血清素的自體受體,數量減少了。結果,這種回饋機制有部分失去功能,而「關掉血清素機器」的訊息也變得相當微弱。突觸前神經元開始再次以正常速率啟動,至少有一陣子如此,而且每次釋放的血清素都比正常值來得多。(^ii,40)
ii但長期而言,血清素的釋放似乎會降到一個低得異常的狀態,至少在特定腦部區域是如此。
當禮來的科學家與其他研究者將氟西汀作用於腦部效果的全貌拼湊起來,他們開始思索,過程中的哪一個部分是抗憂鬱劑特性的來源。精神科醫師長久以來觀察到,抗憂鬱劑要花上二至三週的時間才能「起作用」,因此禮來的研究者於1981年推論,血清素受體的減少(這要花上幾週的時間才會出現)是「與治療反應相關的根本機制」。若是如此,那我們之所以能說藥物有用,是因為它使血清素系統變成較不容易起反應的狀態。可是當研究者發現氟西汀使回饋機制失去部分功能,麥基爾大學的克勞德·德蒙蒂尼(Claudede Montigny)便提出反駁,指出這才是使藥物起作用的原因。這個失能的過程也要花二或三週的時間才會出現,且這使得突觸前神經元開始釋放比正常值更大量的血清素至突觸。在那一刻,氟西汀持續阻斷血清素的移除,神經傳導物質可以真正「堆積」於突觸,並導致「中樞血清素神經傳導的增強」,德蒙蒂尼寫道。(^42)
這就是氟西汀如何改造大腦的科學故事,可能正是這道程序,幫助憂鬱的人們恢復健康並保持健康。唯有用藥成效的文獻能透露是否真是如此。但這藥物顯然並未修復腦中的化學失衡;它其實是起了相反的作用。用藥之前,憂鬱之人並沒有已知的化學失衡。氟西汀破壞了將血清素移開突觸的正常過程,而這又觸發一連串的改變;數週後,血清素路徑便以絕對異常的方式運作。突觸前神經元釋放比平常更多的血清素,血清素回收通道被藥物阻斷;整個系統的回饋循環已部分失能。突觸後神經元變得對血清素「不敏感」,若以機制的觀點來看,血清素系統現在可是被弄得亂七八糟。
禮來的科學家相當明白這個事實。1977年,雷伊·富勒(Ray Fuller)與汪大衛(David Wong)觀察到,既然氟西汀會擾亂血清素路徑,就可用於研究「血清素神經元在不同腦部功能的角色,例如行為、睡眠、調節腦下垂體荷爾蒙釋放、調節溫度、對疼痛的反應等等」。為了進行這類試驗,研究者對動物投予氟西汀,並觀察會有何種功能受損。他們期待病狀出現。事實上,這種類型的研究已經作過了:富勒與汪大衛於1977年報告指出,該藥在大鼠身上激起「典型的過度活動」,並在大鼠與貓的身上皆造成「快速動眼睡眠(REM sleep)受抑制的情況」(^43)
1991年,在一篇發表於《臨床精神醫學期刊》(Journal of Clinical Psychiatry)的論文中,普林斯頓大學的神經科學家貝瑞·雅各(Barry Jacobs)精確表達了關於選擇性血清素回收抑制劑的論點。他寫道:
這些藥物「改變了突觸傳導的水平,使其超出(正常的)環境/生物條件之下所能達到的生理範圍。因此,任何這種情況下產生的行為或生理改變,比較適當的看法應是將其視為病理狀態,而非反映出5-羥基色胺(血清素)正常扮演的生物學角色。」(^44)
1970到1980年代,研究神經抑制劑療效的研究者也描繪出類似的故事。托拉靈與其他標準的抗精神病劑,阻斷了腦中70-90%的第二型多巴胺受體。為了回應這些改變,突觸前神經元開始釋放更多的多巴胺,而突觸後神經元也增加其第二型多巴胺受體密度達30%以上。大腦可說在試圖「補償」藥物的效果,以維持多巴胺路徑一路上的訊息傳遞。然而,大約三週後,這條路徑的回饋機制開始失靈,突觸前神經元開始以不規則的方式啟動,甚至變得靜止不動。正是這種多巴胺路徑「失去功能」,「可能是抗精神病劑作用的基礎」,美國精神醫學會的《精神藥理學教科書》(Textbook of Psychopharmacology)如此解釋。(^45)
超這又是一次神經傳導物質路徑受到藥物影響轉型的故事。數週後,多巴胺路徑的回饋循環部分失能,突觸前神經元釋放出比正常值低的多巴胺,藥物透過阻斷第二型多巴胺受體以妨礙多巴胺的效果,而突觸後神經元則具有異常高密度的受體。此藥並未使腦中的化學恢復正常,反而是擾動了它,若依雅各的推論,某種程度上這可被視為「病理狀態」。
一個理解精神藥物的模式 (p. 118)
今日,史蒂芬‧海曼是哈佛大學的教務長,主要的工作是領導一間大型機構必須應付的諸多政治與行政作業。但他所受的訓練可是要成為一位神經科學家,1996到2001年,當時身為美國國家精神衛生研究院院長的他,寫了一篇兼具紀念性與挑釁的論文,文中總結了人們對於精神科用藥的一切認識。該篇文章的標題是〈開始與適應:一個理解精神藥物如何作用的模式〉(Initiation and Adaptation: A Paradigm for Understanding Psychotropic Drug Action),發表於《美國精神醫學期刊》,文中講述了我們如何理解所有精神藥物作用在大腦的共通方式。(^46 iii)
iii 譯注:抗精神病藥物、精神科藥物,以及精神用藥三個用詞在使用時略有差異。抗精神病藥物(antipsychotics)是指可用以改善精神病(psychosis)症狀的藥物,諸如妄想、幻覺、激動等等。精神科用藥(psychiatric drugs/medications):指的是經過核准、通常由精神科醫師開立處方之藥物,主要包括(1)抗精神病藥物、(2)抗憂鬱劑、(3) 情緒穩定劑、(4)抗焦慮劑/安眠劑/鎮定劑/肌肉鬆弛劑、(5)興奮劑。而無論合法或非法,會影響知覺、情緒、意識等等的藥物,則統稱為精神藥物(psychotropic drugs/medications)
海曼寫道,抗精神病劑、抗憂鬱劑,以及其他的精神藥物,「對神經傳導物質的功能造成擾動」。為了回應上述的改變,大腦便開始進行一系列補償性的適應行為。若藥物阻斷了神經傳導物質(如抗精神病劑所做的那樣),突觸前神經元會換成高速檔並釋放更多神經傳導物質,突觸後神經元則是增加化學傳訊者受體的密度。反過來說,若藥物增加突觸神經傳導物質的濃度(如抗憂鬱劑所做的那樣),則會觸發相反的反應:突觸前神經元減慢啟動的速率,而突觸後神經元減少神經傳導物質受體的密度。在每一個例子中,大腦都在試圖抵消藥物的作用。「這些適應,」海曼解釋道,「是根植於人體內的恆定機制,其存在大抵是要使細胞在面對外界環境變化,或內在環境改變時,仍能保持均衡。」
然而一段時間後,這些補償機制瓦解了。海曼寫道,「長期使用」藥物後來造成了「神經功能本質上的長久改變」。細胞內部訊息傳遞路徑與基因表現的改變,都是長期適應過程的一部分。他的結論是,幾週後,用藥者腦部的運行方式「不管質或量,都與正常狀態不同了」。
海曼的論文寫得優雅,並總結了數十年來令人印象深刻的科學研究所帶來的學問。四十年前,人們發現了托拉靈與其他第一代精神科用藥,科學家對神經元間如何溝通的了解還很少;現在,他們對腦中的神經傳導物質系統與藥物如何在其間作用已有非常詳細的了解。科學告訴我們:用藥物治療前,被診斷為思覺失調症、憂鬱症、以及其他精神疾病的患者並未苦於任何已知的「化學失衡」;然而一旦使用精神科用藥,就像是以某些方式破壞了神經路徑平常的運行,如同海曼觀察到的,患者他(她)的腦袋就開始異常運作。
回到最初 (p. 120)
海曼的論文或許看來相當驚人,但正好可作為科學故事的終章;事實上,這個故事從頭到尾都是一致的。他的論文不應被視為提出了出乎意料的結論,反而該說,精神藥理學一開始的篇章就已如此預測。
一如所見,托拉靈、眠爾通,以及馬西理,都是從為了其他目的所開發的化合物,是從那些可用於手術、或作為對抗傳染病的「神奇子彈」,衍生而來。而後人們發現這些化合物會造成情緒、行為與思考的改變,這些改變似乎對精神病患頗有幫助。本質上,人們認為這些藥物具有有益的副作用。它們擾亂正常的功能,這層理解反映在其最初的命名上。氯丙嗪是「強鎮定劑」,人們並說它能帶來類似額葉切除術的改變。美普巴是「弱鎮定劑」,在動物實驗中顯示,它是一種強烈的肌肉鬆弛劑,並會阻斷受試動物對環境壓力源正常的情緒反應。異菸鹼異丙醯肼是「精神興奮劑」,倘若結核病患在病房跳舞的報導為真,這便是一種能觸發某些類似躁症狀態的藥物。然而,精神醫學接著做的是將這些藥物重新設想為精神疾病的「神奇子彈」,它們被假定為腦中化學失衡的解藥。但這種一半來自於科學,一半來自一廂情願想法的理論,經研究調查後並無法成立。如同海曼所言,精神藥物反倒是擾亂了腦中神經路徑的正常功能。精神醫學對這些新藥的第一印象,原來才是科學上準確的印象。
如今,帶著這種對精神科用藥的了解,我們便有可能提出本書核心的科學問題:長期下來,這些藥物是幫助或傷害了病患?五十年來用藥成效的研究可以告訴我們些什麼?